
Les responsables des unités de retraitement des DMx doivent assurer la veille technologique dans leur domaine. Connaître les modifications des nouvelles versions de normes et des divers documents de référence fait partie de cette responsabilité. L’objectif de cet article est de vous aider à faire une synthèse des nouveautés qui peuvent avoir un impact sur votre activité professionnelle.
ODim
En mai 2021, une nouvelle version de l’ODim a été publiée pour assurer une cohérence avec le nouveau règlement européen sur les DMx (RDM).
Les modifications importantes sont les suivantes :
- Dans l’article 4, il y a les définitions de la maintenance et du retraitement
- Maintenance : des mesures telles que l’entretien, les mises à jour logicielles, les inspections, les réparations, la préparation à la première utilisation et les retraitements en vue de réutiliser, de maintenir ou de rétablir le bon fonctionnement d’un dispositif.
- Retraitement : le procédé dont fait l’objet un dispositif usagé pour en permettre une réutilisation sûre, y compris le nettoyage, la désinfection, la stérilisation et les procédures connexes comme l’emballage, le transport et le stockage, ainsi que l’essai du dispositif usagé et le rétablissement de ses caractéristiques techniques et fonctionnelles en matière de sécurité.
Par conséquent le retraitement fait partie de la maintenance. Il faut donc appliquer l’obligation de maintenance de l’article 49 de la Lpth.
- L’article 20 de l’ancienne version est devenu l’article 71 ci-dessous :
Art. 71 Maintenance
1 Tout professionnel utilisant un dispositif veille à ce que sa maintenance et les tests de maintenance soient réalisés conformément aux exigences légales.
2 La maintenance doit obéir aux principes d’un système de gestion de la qualité et être organisée et documentée adéquatement ; elle se fonde :
A. sur les instructions du fabricant ;
B. sur les risques inhérents au dispositif et à son utilisation.
3 Pour les dispositifs avec fonction de mesure, des procédures de contrôle telles que définies dans l’ordonnance du 15 février 2006 sur les instruments de mesure81 peuvent être prévues.
4 Swissmedic peut formuler et publier des directives concernant la maintenance. Ces directives sont réputées refléter l’état de la science et de la technique.
Les guides publiés par Swissmedic, notamment les Bonnes pratiques suisses de retraitement des dispositifs médicaux font partie de ces directives.
- L’article 19 de l’ancienne version est devenu l’article 72 ci-dessous :
Art. 72 Retraitement
1 Tout professionnel employant un dispositif destiné à être utilisé plusieurs fois veille, avant chaque utilisation, à en vérifier le bon fonctionnement et s’assure que le dispositif a fait l’objet d’un retraitement conforme aux prescriptions selon l’état de la science et de la technique et tenant compte des instructions du fabricant et des exigences en matière d’hygiène.
2 Le retraitement doit être effectué en suivant des procédures adéquates et validées conformément à l’état de la science et de la technique ; leur efficacité dûment attestée, vérifiable et reproductible doit être garantie dans le cadre d’un système de gestion de la qualité.
3 Toute personne retraitant des dispositifs pour des tiers doit :
A. déclarer :
- que le dispositif a été retraité conformément aux instructions du fabricant, ou
- que le dispositif a été retraité selon une procédure propre offrant la même sécurité et la même efficacité que celle prescrite par le fabricant et que cette équivalence a été établie au moyen d’une analyse des risques et d’une procédure de validation ;
B. disposer d’un système de gestion de la qualité approprié, certifié selon des normes nationales ou internationales ;
C. apporter la preuve que le retraitement s’effectue dans des locaux appropriés selon les règles reconnues de la science et de la technique et que les exigences en matière d’hygiène sont respectées;
D. documenter que le dispositif a été retraité conformément à la let. a.
4. La déclaration visée à l’al. 3, let. a, doit mentionner l’identification du dispositif ainsi que le nom et l’adresse de l’entreprise ayant effectué le retraitement.
Le retraitement doit donc être effectué selon les instructions du fabricant et l’état de la science et de la technique.
Les unités de retraitement doivent effectuer leurs activités selon des procédures adéquates et validées.
- L’article 73 précise qu’il est interdit de retraiter des dispositifs à usage unique usagés et de les réutiliser.
- L’article104 précise les délais pour l’apposition de l’IUD (identification unique des dispositifs), soit :
A. pour les dispositifs implantables et les dispositifs de classe III, à partir du 26 mai 2021;
B. pour les dispositifs de classe IIa et IIb, à partir du 26 mai 2023;
C. pour les dispositifs de classe I, à partir du 26 mai 2025;
L’apposition de l’IUD et ses conséquences sur l’unité de retraitement des DMx est une réflexion qu’il faut faire dans chaque établissement.
Normes
- SN EN 17141 : Salles propres et environnement maîtrisé apparentés – Maîtrise de la biocontamination (01.2021)
- Elle remplace la norme SN EN ISO 14698-2
- Le tableau de l’annexe B.3 donnent les limites recommandées pour la surveillance de la contamination microbiologique en environnements propres maîtrisés pendant la fabrication des dispositifs médicaux.
- SN EN ISO 17664-1 et -2 : Traitement de produits de soins de santé – Informations relatives au traitement des dispositifs médicaux à fournir par le fabricant du dispositif (10.2021)
- Partie 1 : Dispositifs médicaux critiques et semi-critiques
- Partie 2 : Dispositifs médicaux non critiques
Cette norme concerne les DMx destinés à être réutilisés et requérant un traitement pour les faire passer de leur état « après utilisation clinique » à l’état « prêt à être réutilisé » et les DMx à usage unique requérant un traitement avant utilisation et destinés à être utilisés dans un état nettoyé et/ou désinfecté. Par exemple, les vis orthopédiques.
La norme a été divisée en deux parties.
- SN EN 285 : Stérilisation – Stérilisateurs à la vapeur d’eau – Grands stérilisateurs (10.2021)
- Dans la définition des gaz non condensables et la vapeur d’eau saturée, la note suivante a été rajoutée : Les exigences relatives à la pénétration de la vapeur d’eau et la définition de « vapeur saturée » constituent actuellement des points de discussion au sein du CEN/TC 102/GT 5 et de l’ISO/TC 198/GT 3.
- 6.4.4.2 – Chaînes de mesure de pression de l’alimentation en vapeur et de la double enveloppe
Les chaînes de mesure de la pression doivent :
- donner des indications en kilopascal ou en bar ;
- avoir une exactitude de ±1,6 % ou plus sur la plage de 0 kPa à 400 kPa ;
- être graduées avec des divisions ne dépassant pas 20 kPa ;
- comporter des moyens permettant un réglage in situ, sans démonter l’instrument.
- 8.1 Pénétration de la vapeur d’eau
NOTE 1 : Chaque procédé de stérilisation à la vapeur d’eau est un évènement unique. Bien qu’un essai de pénétration de vapeur réalisé périodiquement constitue un moyen de contrôle du matériel très utile, des dispositions peuvent être prises pour indiquer qu’une pénétration adéquate de la vapeur d’eau se produit pour chaque cycle.
Cette note irait dans le sens de faire un test de pénétration de vapeur pour chaque cycle de stérilisation.
NOTE 2 : Dans le secteur de la santé, l’utilisation d’instruments à corps creux dotés de longues cavités s’est accrue. La détermination de l’efficacité d’extraction d’air par le biais d’essais portant sur une charge textile peut s’avérer inadéquate pour certains de ces instruments.
Par conséquent, le choix des tests de pénétration de vapeur doit se faire en fonction des charges stérilisées.
SN EN ISO 11138-8 : Stérilisation des produits de santé – Indicateurs biologiques – Partie 8 : Méthode pour la validation d’un temps d’incubation réduit pour un indicateur biologique (08.2021)
- Un temps d’incubation inférieur à 7 j est considéré comme un temps d’incubation réduit, abrégé TIR
- Cette norme concerne la surveillance les procédés de stérilisation à la chaleur humide ou à l’oxyde d’éthylène, mais pas aux vapeurs de peroxyde d’hydrogène.
Si vous utilisez des indicateurs biologiques pour la validation des procédés de stérilisation à la vapeur d’eau, cela vous concerne. Il faudra vous assurer que les indicateurs que vous achetez sont conformes à cette nouvelle partie de la norme SN EN ISO 11138.
SN EN ISO 15883-5 : Laveurs désinfecteurs – Partie 5 : exigences de performance et critère d’essai pour démontrer l’efficacité du nettoyage (10.2021)
- Elle remplace la version 2005
- Dans les termes et définitions, il y en a deux qui sont importantes :
- Propre : Visuellement exempt de souillures et avec des analytes inférieurs aux niveaux spécifiés
- Analyte : substance chimique faisant l’objet d’une analyse chimique
- 4.1.5 : Essai d’efficacité du nettoyage, l’essai de qualification de performance doit être effectué avec la charge souillée par une utilisation clinique dans les conditions les plus défavorables ou, si justifié, avec le produit de substitution.
Il est recommandé d’utiliser du matériel souillé en routine, mais ce n’est pas une obligation. Une charge critique peut aussi être composée de produits de substitution badigeonnés d’une souillure test appropriée.
- 4.2 : Souillures d’essai. La justification de la sélection de la ou des souillures d’essai et de la ou des méthodes de contamination doit être fournie et documentée. La formulation des souillures d’essai peut être choisie ou développée à partir d’une revue de la littérature et de la démonstration de sa pertinence.
- L’annexe A.1 donne des exemples en fonction des types de chirurgie.
Le choix des souillures d’essai utilisées (qualification de performance et contrôles de routine) devra donc être justifié scientifiquement et documenté.
- 4.2.4 : L’efficacité de la méthode d’extraction et de récupération des souillures d’essai ainsi que de détection des analytes doit être validée et spécifiée. La validation de la récupération doit démontrer une capacité à réduire l’analyte en dessous du niveau d’action. Sauf justification contraire, une récupération appropriée exprimée en pourcentage est supérieure à 70 %.
- 4.4 : Critères d’essai d’efficacité du nettoyage. L’efficacité du nettoyage doit être déterminée par un examen visuel et par la détection quantitative des protéines. Les dispositifs médicaux non invasifs ne doivent exiger qu’un examen visuel.
- 4.4.2 : Examen visuel. L’examen visuel doit démontrer l’absence de souillure visible sur l’ensemble des surfaces observables de la ou des charges, au terme de la ou des phases de nettoyage.
NOTE : Les exigences d’examen visuel appropriées peuvent comprendre :
- des consignes de contrôle précises ;
- un éclairage adéquat ;
- des accessoires de contrôle, le cas échéant (par exemple, boroscope avec source lumineuse) ;
- la distance d’examen.
Se reporter à la SN EN 13018 pour de plus amples informations sur l’examen visuel.
Les BPR 2022 recommandent par exemple un éclairage de 1000 Lux pour le contrôle de propreté et de fonctionnalité des instruments.
La norme SN EN 13018 précise que le personnel chargé d’effectuer les contrôles possède une vision satisfaisante conformément à l’EN ISO 9712. De plus, pour l’exécution d’un contrôle visuel général, la vision de loin doit être vérifiée à l’aide d’un optotype normalisé conformément à l’EN ISO 8596, pour un degré d’acuité visuelle de 0,63, mesuré avec au moins un œil, corrigé ou non. La vision doit être vérifiée au moins tous les 12 mois.
A ce jour, c’est un contrôle qui n’est pas fait dans les unités de retraitement des DMx, mais une analyse devrait montrer que c’est une mesure qui réduit les risques de trouver du matériel non conforme chez l’utilisateur (instruments sales, petits trous dans les emballages, etc.).
- 4.4.3 : Critères de dosage. Les critères d’acceptation pour les analytes sont spécifiés en termes à la fois de niveau d’alerte et de niveau d’action.
- Protéines
- Niveau d’alerte ≥ 3 µg/cm2
- Niveau d’action ≥ 6,4 µg/cm2
- L’annexe C décrit les méthodes de dosage.
- Carbone organique total (COT)
- Niveau d’alerte ≥ 6 µg/cm2
- Niveau d’action ≥ 12 µg/cm2
- Glucides
- Niveau d’alerte ≥ 0,9 µg/cm2
- Niveau d’action ≥ 1,8 µg/cm2
- Il y a aussi des valeurs pour l’hémoglobine, l’ATP, les endotoxines
- Protéines
- Annexe B : Évaluation des performances des souillures d’essai protéiniques. Des méthodes d’essai et des appareillages pour tester les souillures sont décrits.
Dans le guide suisse de validation des procédés de lavage et de désinfection des DMx, les valeurs pour les protéines avaient déjà été mentionnées, mais pas pour les autres analytes.
A partir du moment ou des tests sont décrits, il faut demander aux fournisseurs de souillures une attestation de conformité à la présente norme.
SN EN ISO 15223-1 : Dispositifs médicaux – Symboles à utiliser avec les informations à fournir par le fabricant – Partie 1 : Exigences générales (10. 2021)
Les nouveaux symboles publiés dans cette norme et qui concernent notre activité sont les suivants :
Importateur 
Distributeur 
Stérilisé avec du VH2O2 ![]()
SBS unique ![]()
SBS double ![]()
SBS unique avec emballage de protection intérieur 
SBS unique avec emballage de protection extérieur 
Un seul patient plusieurs utilisations 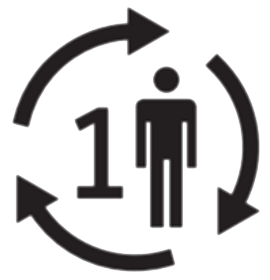
Identifiant unique de dispositif 
SN ISO/TS 16775 : Emballages des dispositifs médicaux stérilisés au stade terminal — Lignes directrices relatives à l’application de l’ISO 11607-1 et l’ISO 11607-2 (11. 2021)
- Cette version existe aussi en français, ce qui n’était pas le cas des versions précédentes. L’annexe B, Recommandations relatives à l’application de la série de normes ISO 11607 dans les établissements de santé est très pratique.
- Vous trouverez les principes de conception d’un système d’emballage, les facteurs à prendre en compte pour la conception et la sélection de l’emballage, des considérations relatives à l’assemblage et un chapitre sur les choix courants de systèmes de barrière stérile.
- Il a aussi un chapitre sur les enveloppes de stérilisation avec les différentes méthodes de pliages (double enveloppe, carré/parallèle).
- Vous trouverez aussi des figures qui décrivent comment effectuer l’ouverture aseptique par l’infirmier/infirmière en bloc stérile
- Il y a un chapitre sur l’évaluation de la stabilité du système de barrière stérile (durée de conservation) et un sur l’aptitude à la présentation aseptique.
- Les exigences de validation (QI, QO, QP et contrôles de routine) pour les procédés de formage, scellage et assemblage pour les procédés suivants sont décrites :
- procédé de scellage: le formage et scellage de sac, sachet et gaine;
- procédé de conditionnement: pliage et fermeture des enveloppes de stérilisation;
- procédé pour le conteneur réutilisable: fermeture des conteneurs réutilisables
- Les annexes J à M donnent des exemples de fiches pour les validations des différentes catégories d’emballages.
- L’annexe N donne un exemple de fiche d’évaluation par les utilisateurs finaux.
La validation des emballages est une exigence des BPR 2022. En attendant la publication d’un guide suisse de validation des systèmes d’emballages pour les procédés de stérilisation, vous pouvez suivre les exigences décrites dans l’annexe B de la norme SN ISO/TS 16775 qui vous donne déjà beaucoup de détails.
Guides
Guide suisse pour le transport des dispositifs médicaux réutilisables souillés et retraités pour les centrales de stérilisation (09.2021)
Bonnes pratiques suisses de retraitement des DMx (01. 2022)

Pour ces deux guides, la SSSH vous recommande d’évaluer vos pratiques, analyser les déviations et les risques et établir un plan d’actions afin de réduire ceux-ci à un niveau acceptable.
Les inspections de Swissmedic se basent sur les BPR 2022 et les guides associés publiés dont fait partie le guide de transport (voir annexe 5 des BPR).
Les versions des normes mentionnées dans cet article peuvent être achetées à la SNV